- Home
- About the Journal
- Peer Review
- Editorial Board
- For Authors
- Reviewer Recognition
- Archive
- Contact
- Impressum
- EWG e.V.
Cite as: Archiv EuroMedica. 2022. 12; 3: e1. DOI 10.35630/2199-885X/2022/12/3.16
Pheochromocytoma, or adrenal paraganglioma, is a neuroendocrine tumor of the adrenal medulla. In the vast majority of cases, pheochromocytoma is a hormonally active tumor with hypersecretion of catecholamines: adrenaline, norepinephrine, dopamine. However, due to the neuroendocrine origin of the tumor, extremely rare cases of ectopic hypersecretion of other peptide hormones by pheochromocytoma have also been described, such as adrenocorticotropic hormone, somatostatin, neuropeptide Y, methenkephalin, and vasoactive interstitial peptide. In the available literature in Russian and English, we found only four cases of calcitonin secretion by pheochromocytoma, the earliest of which was published in 1977.
Examination of a 38-year-old female patient with hypertension (chronically high blood pressure involved sympathoadrenal crises with vegetative dysfunction) revealed a large tumor of the right adrenal gland up to 78 mm. Detailed radiation characteristics of the tumor by ultrasound, computed tomography (CT), magnetic resonance imaging, and positron emission tomography with CT with 18-fluorodeoxyglucose were presented. The results of laboratory tests established hyperproduction of catecholamines by the tumor. In addition, a repeated increase in the level of calcitonin in the blood was revealed, which, in the absence of a genetic study of the RET proto-oncogene, did not allow to exclude the multiple endocrine neoplasia (MEN) type 2. After surgical treatment, the level of calcitonin in the blood decreased to undetectable, which suggested hyperproduction of calcitonin by pheochromocytoma. This hypothesis was confirmed by the results of histological and immunohistochemical studies of the surgical material.
Ectopic production of calcitonin by pheochromocytoma is not typical and is extremely rare, but its development is possible, which should be taken into account when observing its elevated level in patients with pheochromocytoma. Preoperative genetic testing in such cases may significantly reduce the risk of developing medullary thyroid carcinoma in patients without hereditary MEN2.
Keywords: pheochromocytoma, calcitonin, medullary thyroid cancer, multiple endocrine neoplasia type 2, MEN2, calcitonin-producing pheochromocytoma, c-cell hyperplasia.
Pheochromocytoma (PCC, adrenal paraganglioma) is a neuroendocrine tumor of the adrenal medulla with hypersecretion of catecholamines: adrenaline, noradrenaline, dopamine. Due to the neuroendocrine origin of the tumor, rare cases of ectopic hypersecretion of other peptide hormones have been described, such as adrenocorticotropic hormone (ACTH), somatostatin, neuropeptide Y, methenkephalin, and vasoactive interstitial peptide (VIP) [1, 2, 3, 4].
About a third of all PCC/PG patients have hereditary mutations [5], among which an important place is occupied by multiple endocrine neoplasia type 2 (MEN2), associated with a mutation of the RET proto-oncogene. MEN2, with almost 100% penetrance, is accompanied by medullary thyroid carcinoma (MTC), which biochemical marker is calcitonin.
We present a rare clinical case of calcitonin hypersecretion by pheochromocytoma without MTC.
A 38-year-old female patient was hospitalized at Saint Petersburg State University Hospital with complaints of increased blood pressure (BP) crises up to 180/100 mm Hg. under constantly elevated BP up to 140-150/90-95 mm Hg., accompanied by throbbing headaches in the occipital region, palpitations, profuse sweating, fear, a feeling of internal trembling, shortness of breath, 2-3 times a week.
Over the course of two years, she repeatedly consulted a general practitioner and a cardiologist, but the change in antihypertensive therapy did not bring improvement. During the last year, there was an increase in fasting plasma glucose to 6.2 mmol/l with 6% glycated hemoglobin.
Screening ultrasound of the abdominal organs revealed for the first time an isoechogenic formation with clear contours, a homogeneous structure, dimensions 89x67x66 mm in the subhepatic space on the right. Contrast-enhanced abdominal MRI showed that a round-shaped mass of 72x74x68 mm originates from the right adrenal gland, with a heterogeneous MR signal in the center due to the presence of areas of necrosis, intensively heterogeneously accumulating contrast agent (CA). Whole-body PET/CT with 18F-FDG in the projection of the adrenal gland revealed a round solid mass of 78x70x76 mm with diffuse uneven hypermetabolism of 18F-FDG with SUV max=5.98. No other foci of imaging agent were found.
An outpatient examination revealed an increase in the levels of free metanephrine and free normetanephrine in daily urine up to 316 mcg/day (18-277 mcg/day) and 9164 mcg/day (42-423 mcg/day). Plasma aldosterone was 91.7 pg/ml (25.2-392), and renin was 10.7 µIU/ml (4.4-46.1). Excretion of cortisol in daily urine: 107 nmol / day (up to 485.6). Overnight suppression test with 1 mg dexamethasone showed serum cortisol 0.8 mcg/dL. Pheochromocytoma was diagnosed, and surgical treatment was recommended. Analyzes to exclude MEN 2: Ca++ 1.22 mmol/l (1.12-1.32), parathyroid hormone 5.4 pmol/l (1.6-6.9), calcitonin 28.35 pg/ml (0-6.4), repeated in another laboratory - 31 pg/ml (0-5). In the hospital: calcitonin 12.4 pg/ml (0-5), CEA 2.6 ng/ml (0-3.8), chromogranin A 139 µg/l (0-100). Ultrasound of the thyroid gland revealed no pathology.
CT scan of the thoracic and abdominal organs with contrast: thoracic organs without pathology. In the right adrenal gland, a neoplasm of 75 mm, with even clear contours, tightly adjacent to the lower surface of the right lobe of the liver, to the anterior surface of the upper part of the right kidney, to the inferior vena cava, with a density of +25HU, in the arterial phase up to +116HU, in the venous phase up to + 105HU, 10 minutes after administration of imaging agent +60 HU. Large vessels are seen in the structure of the neoplasm. Fig. 1.
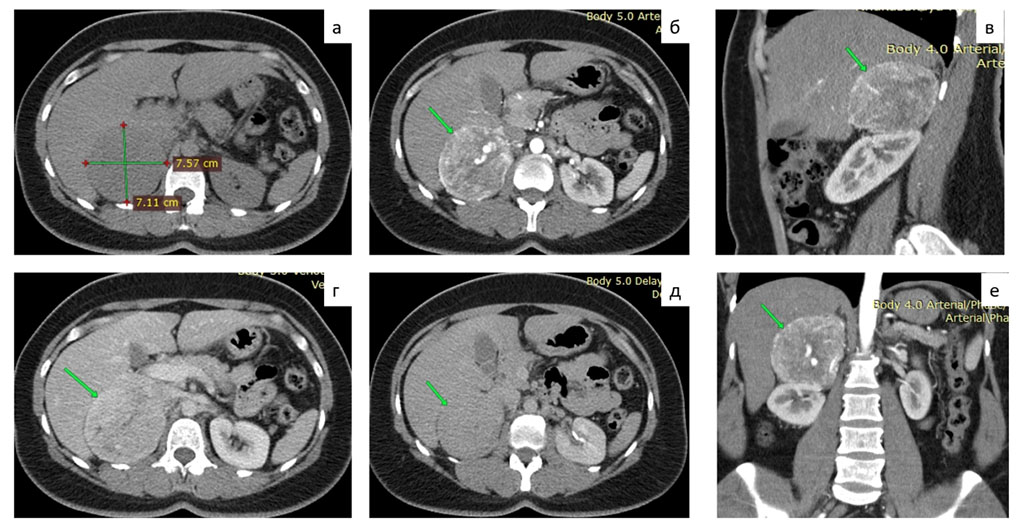
Figure 1. MSCT of the abdomen with contrast: a - native phase, b, c, f - arterial phase, d - venous phase, e - delayed phase.
The patient received preoperative preparation (doxazosin 8 mg per day, bisoprolol 10 mg per day) for 3 weeks, after which a right single-port retroperitoneoscopic adrenalectomy was performed. Fig. 2.
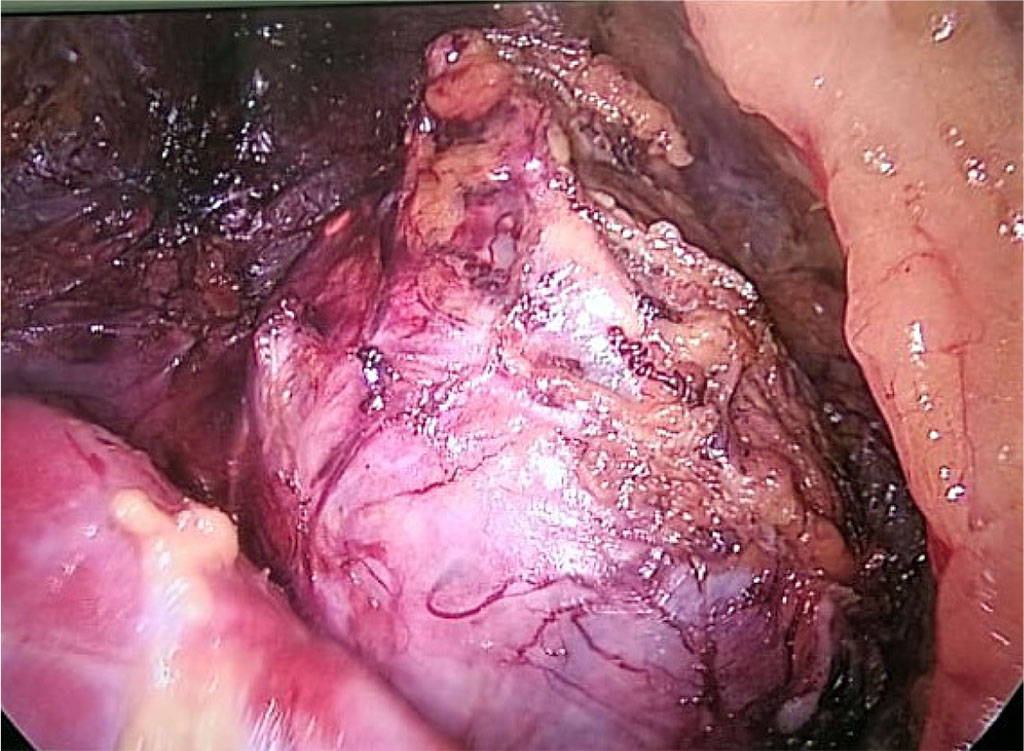
Figure 2. Intraoperative image of a pheochromocytoma of the right adrenal gland
After the operation, a stimulation test was performed with intravenous injection of 20 ml of 10% calcium gluconate, with the determination of the level of calcitonin in the blood before, as well as 1, 2 and 5 minutes after the administration of the drug. In all studied blood samples, the value of calcitonin was less than 1 ng/ml. Mutations of the RET proto-oncogene were not found. Hyperproduction of calcitonin by the removed tumor of the adrenal gland was suspected.
Histology with immunohistochemistry: tumor of the adrenal gland of the alveolar-nested pattern, with signs of neuroendocrine differentiation, with pronounced cell-nuclear cell polymorphism, with extensive, weak granular cytoplasm, without signs of capsular and vascular invasion. Tumor cells intensively express Chromogranin A; few cells focally intensively express Calcitonin and do not express Melan A. Ki-67 1-2%. Fig. 3.
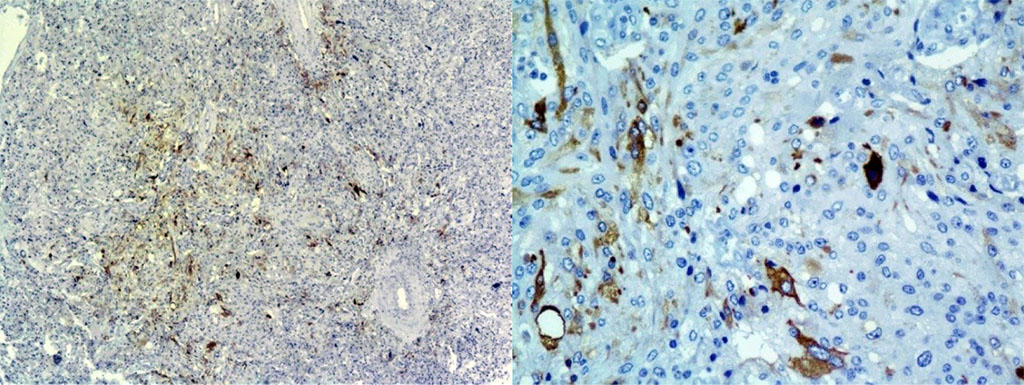
Figure 3. IHC study of an adrenal tumor: Calcitonin+ pheochromocytoma cells, focal focal expression: x40; x200
In the described case, a patient with pheochromocytoma had an elevated level of calcitonin, while ultrasound showed no changes in the thyroid gland, which cast doubt on the presence of MEN2. Other diseases were excluded: hyperparathyroidism, alcoholic cirrhosis, thyroiditis, Paget's disease, breast cancer, pancreatitis, megaloblastic anemia, leukemia. A rare cause of hyperproduction of calcitonin can be neuroendocrine tumors (NETs) outside the thyroid gland - more often in the pancreas [6], less often in the lungs [7], the presence of which is excluded according to CT scans of the chest and abdomen. A low level of calcitonin can occur with C-cell hyperplasia of the thyroid gland, but the normalization of its level after adrenalectomy testified in favor of the production of calcitonin by pheochromocytoma, which was confirmed by IHC.
A similar case was described by Hearth H. and Edis A.J. (1979), in which hypersecretion of calcitonin by the tumor was confirmed by blood sampling from the peripheral and adrenal veins, as well as a decrease in its level after adrenalectomy [8]. A 55-year-old female patient of Kanamori A. et al. (1977) had a history of elevated calcitonin which led to thyroidectomy as the second stage after removal of the pheochromocytoma, despite a drop in its level after the first operation. Histologically, neither thyroid cancer nor C-cell hyperplasia was detected [9].
In the available literature, we found only four cases describing the secretion of calcitonin in PCC [1, 3, 9, 10]. In a review by Kirkby-Bott J. et al. (2012) only three cases are listed, one of which is in Japanese [10]. Ectopic production of calcitonin by adrenal PCC is not typical and is extremely rare, but its development is possible, which should be taken into account when observing its elevated level in patients with PCC, especially in differential diagnosis with MEN2.
Therefore, preoperative genetic testing in patients with pheochromocytoma is recommended. It greatly facilitates the diagnostic search and significantly reduces the risk of developing medullary thyroid carcinoma in patients without hereditary MEN2.
The authors declare the absence of obvious and potential conflict of interest related to the publication of this article.

|
||