- Home
- About the Journal
- Peer Review
- Editorial Board
- For Authors
- Reviewer Recognition
- Archive
- Contact
- Impressum
- EWG e.V.
Purpose: To report a novel case of FECD in a neonate with myotonic dystrophy type 1.
Observations: A 32-week neonate admitted to the NICU (Neonatal Intensive Care Unit). Patient was suspected to have myotonic dystrophy, which was confirmed with DMPK PCR (Polymerase Chain Reaction) and Southern Blot Analysis. Ocular evaluation revealed iris transillumination, bilateral hazy cornea, intraocular pressure (IOP) of 18mmHg for right eye and 22mmHg for left eye, and the centre cornea thickness (CCT) was 862 microns and 887 microns for right and left eye, respectively. The thick and hazy corneas were suggestive of Fuchs’ endothelial corneal dystrophy (FECD).
Conclusions: This may be the first reported case of FECD in neonate with DM1. Knowledge that FECD may present in neonates with DM1 allows for earlier identification and treatment, resulting in decreased vision loss from FECD.
Keywords: myotonic, dystrophy, fuch’s, endothelial
The congenital type 1 form of myotonic dystrophy (DM1) is an autosomal dominant genetic disorder caused by trinucleotide repeat expansion of CTG (cytosine-thymine-guanine) in the DMPK (dystrophia myotonica protein kinase) gene on chromosome 19q 13.3. Genetic research into the molecular pathogenesis of DM has resulted in it becoming the prototypical disease for RNA gain-of-function toxicity [1].
Fuchs endothelial corneal dystrophy (FECD) is a non-inflammatory, sporadic or autosomal dominant, dystrophy involving the endothelial layer of the cornea, and approximately 50% of clinical cases have a familial history [2]. It is bilateral, slowly progressive, often asymmetric corneal disease presenting with loss of endothelial cells and development of guttata and subsequent corneal stroma hydration [3]. The strongest association identified has been with expansion of the CTG18.1 trinucleotide repeat in transcription factor 4 (TCF4) [3]. The current understanding of the disease indicates that RNA toxicity due to an intronic (noncoding) CTG trinucleotide repeat (TNR) expansion in TCF4 may contribute to the pathogenesis of FECD [5].
Gattey and colleagues described typical clinical and histologic features of FECD in four adults from three families with established DM1 [1]. A relationship between DM1 and FECD because the mechanism of CTG repeat induced RNA toxicity may be partially independent of chromosomal location of the TNR expansion within the genome [5]. Winkler et al. suggested that patients with DM1 are prone to developing FECD because of the common pathophysiologic mechanism of RNA toxicity between FECD and DM1 [5]. This is further supported by Mootha and colleagues, who reported that mutant expansions in DMPK and TCF4 share important similarities, including nuclear foci that contain expanded CUG repeats, association of foci with MBNL1 protein, and an ability to cause FECD [6]. All cases reported on association between DM1 and FECD are in adults, we believe this is the first reported case of FECD in a neonate with DM1.
A patient, gestational age 32+5 weeks, Birth weight 1692g was admitted to the NICU (Neonatal Intensive Care Unit). Patient’s birth weight was 1692g. Patient delivered via caesarean section. Patient systemically presented with restrictive lung diseases, diaphragmatic eventration, and hypotonia. Patient was suspected to have severe myotonic dystrophy, which was confirmed with DMPK PCR (myotonic dystrophy protein kinase polymerase chain reaction) and Southern Blot Analysis. The PCR analysis indicated a CTG repeat of 20, TPPCR (Triplet-Repeat Primed Polymerase Chain Reaction) analysis showed detection of expansion, and the size of the CTG repeat was estimated using Southern blot analysis, which demonstrated a large CTG repeat expansion of 1700. Patient was diagnosed with myotonic dystrophy type 1. Patient’s mother was clinically observed to be expressing symptoms of myotonic dystrophy. The patient’s eye examination showed bilateral diffuse stromal fine haze cornea, iris transillumination, intraocular pressure (IOP) of 18mmHg for right eye and 22mmHg for left eye, and the centre cornea thickness (CCT) was 862 microns and 887 microns for right and left eye, respectively. The patient remained in NICU and entirely dependent on respiratory support. Patient is currently managed with 5% sodium chloride four times a day.
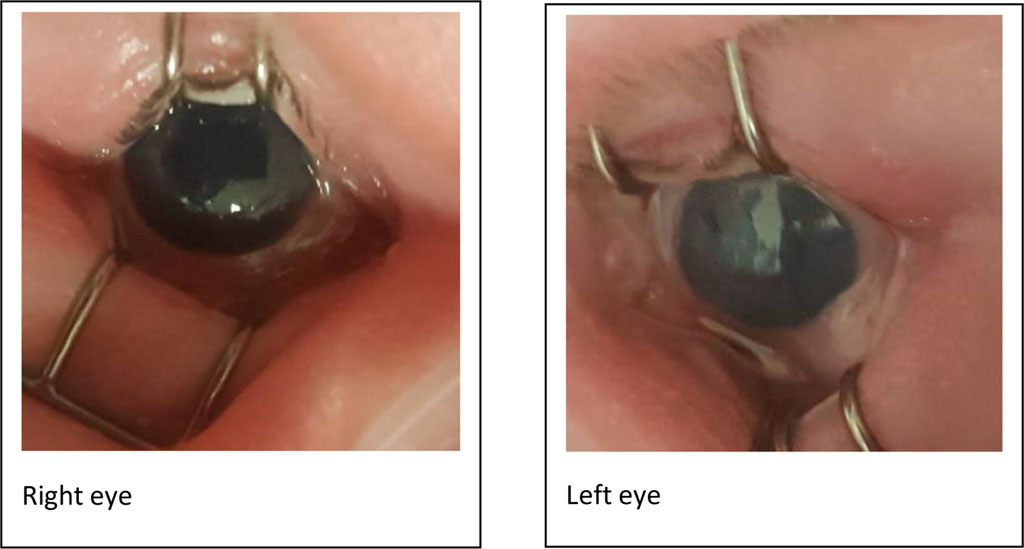
Figure 1. Photo showing hazy cornea (Left > right)
The association of FECD with myotonic dystrophy in our findings was similar to previous case studies by Mootha et al. [6]. The study Included 13 consecutive unrelated DM1 patients for FECD, Six of thirteen DM1 patients (46%) had slit lamp and specular microscopic findings of FECD [6]. The study further demonstrated that DM1 patients are at risk for FECD and established a connection between two repeat expansion disorders converging upon RNA-MBNL1 foci and FECD [6]. Rare heterozygous mutations in collagen, type VIII, alpha 2 gene (COL8A2, MIM 120252) cause an early-onset corneal endothelial dystrophy. A small portion of adult-onset FECD cases are caused by other genes including solute carrier family 4, sodium borate transporter member 11 (SLC4A11, MIM 610206), transcription factor 8 (TCF8, MIM 189909), lipoxygenase homology domains 1 (LOXHD1, MIM 613267), and ATP/GTP binding protein-like (AGBL1, MIM 615523) [6]. Outcomes from the latter suggest that the triplet expansions in both DMPK and TCF4 may cause the same corneal endothelial tissue phenotype of FECD through shared molecular mechanisms that are triggered by toxic gain-of-function RNA [6]. With genetic abnormality associated with most cases of FECD identified, there is a better understanding of how the diseases might develop and this offers the potential for non-surgical therapy in the future. This case suggests that such therapies may need to be initiated at a much earlier stage.
The current literature strongly supports that, adults with DM1 are prone to developing FECD. This case of FECD in a neonate with a known diagnosis of DM1 is the first to be reported, and this may suggest FECD in DM1 patients can be diagnosed at an earlier age. It may also translate into new opportunities for prompt or preliminary identification and treatment of FECD.
The authors declare that they have NO affiliations with or involvement in any organization or entity with any financial interest in the subject matter or materials discussed in this manuscript.

|
||